Defining Bioregions
This method was described in Brocklehurst, N., & Fröbisch, J. 2018. The definition of bioregions in palaeontological studies of diversity and biogeography affects interpretations: Palaeozoic tetrapods as a case study. Frontiers in Earth Sciences (6): 200. See this paper for references and more detail on the concepts
In studies of alpha diversity (local species richness) and beta diversity (faunal provinciality), it is probably not surprising that the definition of the regions will affect the results you see. It's not that there is a particular set of regions that you should use; rather that different patterns will be observed at different spatial scales. I want to emphasise this is not a problem. The impact of different barriers and environmental controls on the dispersal and range-size of species are expected to, and have been shown to, have different effects depending on what scale you are looking at. Large physical barriers like mountain ranges and sea barriers are unlikely to affect diversity and dispersal patterns at local scales, and local habitat variations are not going to control the dispersal of species between large landmasses. The point I want to make is that the spatial scale at which you define the regions in your study of alpha or beta diversity need to be relevant to the question you are asking and should be considered when making conclusions.
With this in mind, it is unfortunate that palaeontologists have often been a bit inconsistent and arbitrary in their definitions. Alpha and beta diversity has been calculated between localities, formations, basins, and continents, and rarely is there a justification for the particular set of regions used or an acknowledgement that the results obtained will not be directly comparable to those obtained at other spatial scales.
For inspiration on how to better define the bioregions used in palaeontological diversity studies, I’ve turned to the neontologists, who have been defining bioregions rather more rigorously since the 1800s. The biogeographic realms of Philip Sclater and Alfred Russell Wallace, and subsequent updates, were defined as areas of endemicity; they were defined by the taxa under study. This not only ensures that the boundaries between the regions are biologically meaningful, but that the regions are relevant to the specific clade under study.
There are a few issues with bringing these principals into the palaeontological realm. We aren’t dealing with species ranges, but points on a map. The solution to this is to use two hierarchical cluster analyses to group the points. The first groups them based of their geographic distances from each other, the second based on their taxonomic distances (using whatever metric you prefer). Clusters shared between the two will represent continuous geographic areas of endemicity and are suitable for use in diversity studies. The second issue we as palaeontologists face is that the bioregions change through time; barriers will build up and break down through climate changes and movement of continents. So, if we can’t standardise the bioregions used in each time period, we instead need to standardise the spatial scale at which they are defined. We do this by choosing a consistent geographic cluster node height and grouping all localities below that height into their bioregions i.e. if the height chosen is 1000km, all localities within 1000 km of each other will be grouped into their bioregions, if they form a unique taxonomic cluster.
The function I've written (see link in tutorial) takes two distance metrics (one with geographical distances between localities or formations, and one with taxonomic distances), and identifies which localities are grouped together at which cluster node heights. I have an example dataset, and this tutorial will show you how to get the distance matrices and then analyse them.
1. If not yet installed, install the packages vegan, letsR, paleotree and phangorn*
*NOTE: only phangorn and paleotree is actually required for the define.bioregions() function, since this function treats the cluster dendrograms as phylogenies (purely for ease of coding). The other two packages are required for the example code presented here.
2. Read in these packages
library(vegan)
library(letsR)
library(phangorn)
library(paleotree)
3. Download and read in the define.bioregions() function.
4. Download the example file and place it in your working directory; you can check what your working directory is with the line:
getwd()
5. Load data and store as an object called “dataset”. The data needs to be a matrix of presences/absences or abundances, depending on what distance method is to be used with taxa in rows and localities in columns. The last two rows should the longitude and latitude of the localities. The example file you downloaded before can be read into R using the following line:
dataset<-read.csv("example2.csv",row.name=1,header=T)
6. Separate the taxonomic data from the geographic coordinates into two objects: tax.data (the presence/absence data) and geo.data (the coordinates)
tax.data<-dataset[1:(nrow(dataset)-2),]
geo.data<-dataset[(nrow(dataset)-1): nrow(dataset),]
7. Calculate the taxonomic distances between localities. The package “vegan” includes a variety of distance metrics. Here the Bray Curtis metric is used (purely as example, not for personal preference)
tax.dist<- vegdist(t(tax.data),method="bray")
8. Calculate the geographic distances between the localities. The package “letsR” contains a function to do this:
geo.dist<-lets.distmat(t(geo.data))
9. Run the define.bioregions() function**. There are four arguments:
· tax.dist – the taxonomic distance matrix (object of class dist)
· geo.dist – the geographic distance matrix (object of class dist)
· method – the agglomeration method used in the hierarchical cluster analysis (same options as in the hclust() function)
· plot – logical (TRUE or FALSE). If TRUE, will plot the geographic and taxonomic cluster dendrograms.
bioregions<-define.bioregions(tax.dist=tax.dist,geo.dist=geo.dist,method="average",plot=T)
**NOTE: a warning message will always come up: "Root age not given; treating tree as if latest tip was at modern day (time=0)". This is not a problem; its an artefact of an internal paleotree function I use, and I don't know how to stop it sending the message.
10. Examine output by typing bioregions into the R console. The function outputs a list of length two, with names:
· $bioregions – a list of variable length. Each level of the list contains a vector, with each element named after a locality in the dataset. Each element is a number. Those localities with the same number are grouped into a bioregion
· $height.ranges – a list of the same length as $bioregions. Gives the range of cluster node heights between which the groupings indicated in $bioregions apply (i.e. the spatial scale to be used for any analysis).
In studies of alpha diversity (local species richness) and beta diversity (faunal provinciality), it is probably not surprising that the definition of the regions will affect the results you see. It's not that there is a particular set of regions that you should use; rather that different patterns will be observed at different spatial scales. I want to emphasise this is not a problem. The impact of different barriers and environmental controls on the dispersal and range-size of species are expected to, and have been shown to, have different effects depending on what scale you are looking at. Large physical barriers like mountain ranges and sea barriers are unlikely to affect diversity and dispersal patterns at local scales, and local habitat variations are not going to control the dispersal of species between large landmasses. The point I want to make is that the spatial scale at which you define the regions in your study of alpha or beta diversity need to be relevant to the question you are asking and should be considered when making conclusions.
With this in mind, it is unfortunate that palaeontologists have often been a bit inconsistent and arbitrary in their definitions. Alpha and beta diversity has been calculated between localities, formations, basins, and continents, and rarely is there a justification for the particular set of regions used or an acknowledgement that the results obtained will not be directly comparable to those obtained at other spatial scales.
For inspiration on how to better define the bioregions used in palaeontological diversity studies, I’ve turned to the neontologists, who have been defining bioregions rather more rigorously since the 1800s. The biogeographic realms of Philip Sclater and Alfred Russell Wallace, and subsequent updates, were defined as areas of endemicity; they were defined by the taxa under study. This not only ensures that the boundaries between the regions are biologically meaningful, but that the regions are relevant to the specific clade under study.
There are a few issues with bringing these principals into the palaeontological realm. We aren’t dealing with species ranges, but points on a map. The solution to this is to use two hierarchical cluster analyses to group the points. The first groups them based of their geographic distances from each other, the second based on their taxonomic distances (using whatever metric you prefer). Clusters shared between the two will represent continuous geographic areas of endemicity and are suitable for use in diversity studies. The second issue we as palaeontologists face is that the bioregions change through time; barriers will build up and break down through climate changes and movement of continents. So, if we can’t standardise the bioregions used in each time period, we instead need to standardise the spatial scale at which they are defined. We do this by choosing a consistent geographic cluster node height and grouping all localities below that height into their bioregions i.e. if the height chosen is 1000km, all localities within 1000 km of each other will be grouped into their bioregions, if they form a unique taxonomic cluster.
The function I've written (see link in tutorial) takes two distance metrics (one with geographical distances between localities or formations, and one with taxonomic distances), and identifies which localities are grouped together at which cluster node heights. I have an example dataset, and this tutorial will show you how to get the distance matrices and then analyse them.
1. If not yet installed, install the packages vegan, letsR, paleotree and phangorn*
*NOTE: only phangorn and paleotree is actually required for the define.bioregions() function, since this function treats the cluster dendrograms as phylogenies (purely for ease of coding). The other two packages are required for the example code presented here.
2. Read in these packages
library(vegan)
library(letsR)
library(phangorn)
library(paleotree)
3. Download and read in the define.bioregions() function.
4. Download the example file and place it in your working directory; you can check what your working directory is with the line:
getwd()
5. Load data and store as an object called “dataset”. The data needs to be a matrix of presences/absences or abundances, depending on what distance method is to be used with taxa in rows and localities in columns. The last two rows should the longitude and latitude of the localities. The example file you downloaded before can be read into R using the following line:
dataset<-read.csv("example2.csv",row.name=1,header=T)
6. Separate the taxonomic data from the geographic coordinates into two objects: tax.data (the presence/absence data) and geo.data (the coordinates)
tax.data<-dataset[1:(nrow(dataset)-2),]
geo.data<-dataset[(nrow(dataset)-1): nrow(dataset),]
7. Calculate the taxonomic distances between localities. The package “vegan” includes a variety of distance metrics. Here the Bray Curtis metric is used (purely as example, not for personal preference)
tax.dist<- vegdist(t(tax.data),method="bray")
8. Calculate the geographic distances between the localities. The package “letsR” contains a function to do this:
geo.dist<-lets.distmat(t(geo.data))
9. Run the define.bioregions() function**. There are four arguments:
· tax.dist – the taxonomic distance matrix (object of class dist)
· geo.dist – the geographic distance matrix (object of class dist)
· method – the agglomeration method used in the hierarchical cluster analysis (same options as in the hclust() function)
· plot – logical (TRUE or FALSE). If TRUE, will plot the geographic and taxonomic cluster dendrograms.
bioregions<-define.bioregions(tax.dist=tax.dist,geo.dist=geo.dist,method="average",plot=T)
**NOTE: a warning message will always come up: "Root age not given; treating tree as if latest tip was at modern day (time=0)". This is not a problem; its an artefact of an internal paleotree function I use, and I don't know how to stop it sending the message.
10. Examine output by typing bioregions into the R console. The function outputs a list of length two, with names:
· $bioregions – a list of variable length. Each level of the list contains a vector, with each element named after a locality in the dataset. Each element is a number. Those localities with the same number are grouped into a bioregion
· $height.ranges – a list of the same length as $bioregions. Gives the range of cluster node heights between which the groupings indicated in $bioregions apply (i.e. the spatial scale to be used for any analysis).
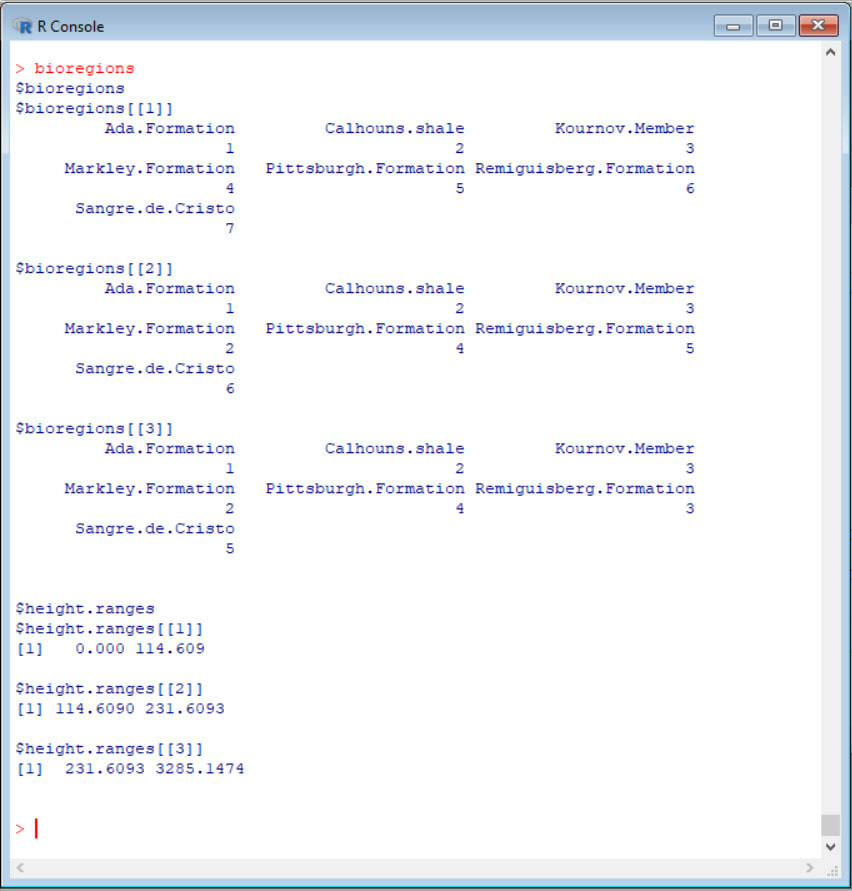
11. So, in practical terms, how would one use this output. Well, lets say you had decided you wanted to count the number of species in each region (alpha diversity) and you decided you were defining your regions at a spatial scale of 200km (every locality within 200km of each other are grouped into their bioregions if they form a taxonomic cluster. If we look at bioregions$height.ranges, we see it is level 2 that we want; that level applies to node heights between 114.609 and 231.6093km. So we now look at the bioregions defined at that node height : bioregions$bioregions[[2]]. We see there that both the Calhouns shale and Markley formation are assigned the same number; they should therefore be grouped together in the single bioregion, and the species in both counted towards the diversity of that bioregion. All other localities are given different numbers and so should each be treated as their own regions.
